Overview
GROMACS is a freely available software package designed for high-performance molecular dynamics simulations and subsequent analysis of output data. GROMACS is an adaptable tool for simulating the Newtonian equations of motion for systems containing hundreds to millions of particles, or molecular dynamics. Since GROMACS calculates nonbonded interactions, which typically dominate simulations, very quickly, it is primarily used for biochemical molecules with complex bonded interactions, such as proteins, lipids, and nucleic acids. However, many researchers also use it for nonbiological systems such as fluid dynamics and polymers.
This article demonstrates how to build GROMACS using the Intel® oneAPI DPC++/C++ Compiler and enable SYCL* support on Intel GPUs. Starting from GROMACS 2021, SYCL compatibility has been incorporated. The current SYCL implementation can be compiled with the Intel oneAPI DPC++/C++ Compiler for Intel GPUs. SYCL is a modern portable heterogeneous acceleration API with multiple implementations targeting various hardware platforms. GROMACS can be used with one of the SYCL compilers or runtimes using Intel® oneAPI DPC++ Compiler (both OpenCL™ and Level Zero back ends) to target the Intel GPU hardware. GROMACS uses SYCL for GPU acceleration on Intel GPUs.
Prerequisites
Before you start building:
- Ensure CMake* platform version 3.18.4 or later is installed.
- GROMACS is compatible with various operating systems (Linux*, macOS*, and Microsoft Windows*) and various hardware architectures. For more information, refer to the Installation Guide for GROMACS.
- As we are building on Intel GPUs, check whether you meet all the system requirements for the Intel oneAPI DPC++/C++ Compiler and Intel® oneAPI Math Kernel Library (oneMKL).
- Install the Intel oneAPI DPC++/C++ Compiler and Intel oneAPI Math Kernel Library (oneMKL), which are available in the Intel® oneAPI Base Toolkit and Intel® HPC Toolkit. It is also available as a stand-alone product.
Build GROMACS on Linux*
- To use the Intel oneAPI DPC++/C++ Compiler, source the Intel oneAPI environment variables using the setvars.sh script.
source /opt/intel/oneapi/setvars.sh - Download the GROMACS source code. Here we have downloaded GROMACS version 2023.3.
Refer to the current source code in the GROMACS documentation.wget https://ftp.gromacs.org/gromacs/gromacs-2023.3.tar.gz
- After downloading, extract the GROMACS source code from the tarball.
xfz gromacs-2023.3.tar.gz - Navigate to the newly extracted GROMACS source directory, create a build directory, and then navigate to it.
gromacs-2023.3mk dir build cd build - Use the CMake tool to configure GROMACS with specific options. The following command sets various build options, such as using oneMKL for fast Fourier transform (FFT) calculations, enabling SYCL for GPU support, and specifying Intel® C and C++ compilers. Command-line parameters are explained as follows.
cmake .. -DGMX_BUILD_OWN_FFTW=ON -DREGRESSIONTEST_DOWNLOAD=ON -DCMAKE_C_COMPILER=icx -DCMAKE_CXX_COMPILER=icpx -DGMX_GPU=SYCL -DGMX_FFT_LIBRARY=mkl
- DCMAKE_C_COMPILER=Intel oneAPI DPC++ Compiler (or the environment variable CC). We are using Intel oneAPI DPC++ Compiler.
- DCMAKE_CXX_COMPILER=Intel oneAPI DPC++/C++ Compiler (or the environment variable CXX). We are using Intel oneAPI DPC++/C++ Compiler.
The Intel C Compiler Classic is no longer supported in GROMACS. We are using Intel compilers for oneAPI. - DGMX_GPU=SYCL to build with SYCL support enabled (using Intel oneAPI DPC++ Compiler by default). We are choosing SYCL back end for GPU offloading.
- DGMX_FFT_LIBRARY=oneMKL. We are using oneMKL. We can also select FFTW 3, MKL, or FFTPACK libraries for fast Fourier transform (FFT) support.
Note When compiling for Intel® Data Center GPU Max Series, it is recommended to pass additional flags for compatibility and improved performance:
cmake .. -DCMAKE_C_COMPILER=icx -DCMAKE_CXX_COMPILER=icpx -DGMX_GPU=SYCL -DGMX_GPU_NB_NUM_CLUSTER_PER_CELL_X=1 -DGMX_GPU_NB_CLUSTER_SIZE=8
- Once the configuration is complete, you can proceed to build GROMACS. To ensure that the GROMACS build is working correctly, run tests.
make make check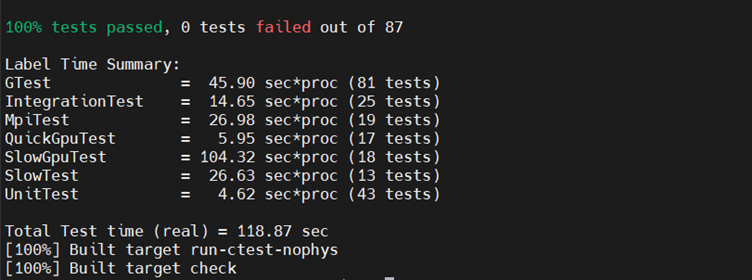
- Once all the tests are passed, you can install GROMACS system-wide using the following command.
sudo make install - Source GMXRC to get access to GROMACS.
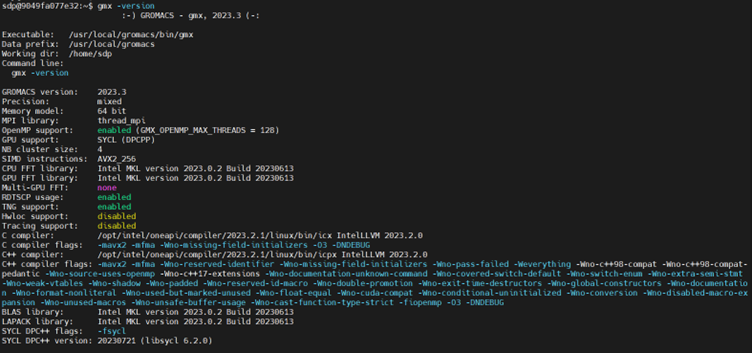
/usr/local/gromacs/bin/GMXRC - To verify whether GROMACS is perfectly installed and configured with SYCL and oneMKL support, run the following command.
This command displays details about the installed version of GROMACS, indicating that it was built using the Intel oneAPI DPC++/C++ Compiler with GPU support enabled through SYCL and oneMKL support enabled.gmx -version
If the output returns the phrase, gmx: command not found, then you must find where your version of GROMACS is installed. In the default case, the binaries are in /usr/local/gromacs/bin path.