Introducing Intel® Select Solutions for Genomics Analytics
This guide focuses on software configuration recommendations for users who are already familiar with the Intel® Select Solutions for Genomics Analytics. The following guide contains the hardware configuration for the HPC clusters that are used to run this software: HPC Cluster Tuning on 3rd Generation Intel® Xeon® Scalable Processors. However, please carefully consider all of these settings based on your specific scenarios. Intel® Select Solutions for Genomics Analytics can be deployed in multiple ways and this is a reference to one use-case.
Genomics analysis is accomplished using a suite of software tools including the following:
- Genomic Analysis Toolkit (GATK) Version 4.1.9.0 is used for variant discovery. GATK is the industry standard for identifying SNPs and indels in germline DNA and RNAseq data. It includes utilities to perform tasks such as processing and quality control of high-throughput sequencing data.
- Cromwell Version 52 is a Workflow Management System for scientific workflows. These workflows are described in the Workflow Description Language (WDL). Cromwell tracks workflows and stores the output of tasks in a MariaDB database.
- Burrows-Wheeler Aligner (BWA) Version 0.7.17 is used for mapping low-divergent sequences against a large reference genome, such as the human genome.
- Picard Version 2.23.8 is set of tools used to manipulate high-throughput sequencing (HTS) data stored in various file formats such as: SAM, BAM, CRAM, or VCF.
- VerifyBAMID2 works with Samtools Version 1.9 to verify whether sample genomic data matches previously known genotypes. It can also detect sample contamination.
- Samtools Version 1.9 are used to interact with high-throughput sequencing data including: reading, writing, editing, indexing, or viewing data stored in the SAM, BAM, or CRAM format. It is also used for reading or writing BCF2, VCF, or gVCF files and for calling, filtering, or summarizing SNP and short indel sequence variants.
- 20K Throughput Run is a simple and quick benchmark test used to ensure that the most important functions of your HPC cluster are configured correctly.
- Optional: Intel® System Configuration Utility (SYSCFG) Version 14.2 Build 8 command-line utility can be used to save and to restore system BIOS and management firmware settings.
These prerequisites are required:
- Git is a version control system for tracking changes.
- Either Java 8 or the Java Runtime Environment (JRE) plus the Software Developers Kit (SDK) 1.8 is required to run Cromwell.
- Python Version 2.6 or greater is required for the gatk-launch. Newer tools and workflows require Python 3.6.2 along with other Python packages. Use the Conda package manager to manage your environment and dependencies.
- Slurm Workload Manager provides a means of scheduling jobs and allocating cluster resources to those jobs. For a detailed description of Slurm, please visit: https://slurm.schedmd.com/documentation.html.
- sbt is required to compile Cromwell.
- MariaDB is used by Cromwell for persistent storage.
- R, Rscript, gsalib, ggplot2, reshape, and gplots are used by GATK to produce plots.
3rd Generation Intel® Xeon® Scalable processors deliver platforms with built-in AI acceleration that can be optimized for many workloads. They provide a performance foundation to help speed data’s transformative impact, from a multi-cloud environment to the intelligent edge and back. Improvements of particular interest to this workload are:
- Enhanced Performance
- Increased DDR4 Memory Speed & Capacity
- Intel® Advanced Vector Extensions
- Intel® Genomics Kernel Library with AVX-512
Architecture
Scientists use tools such as, the Genomic Analysis Toolkit, along with WDL scripts to process DNA and RNA data. Cromwell is used to manage the workflow. SLURM is used to schedule jobs and to allocate cluster resources to those jobs. This diagram shows the software and hardware used in the Intel® Select Solutions for Genomics Analytics.
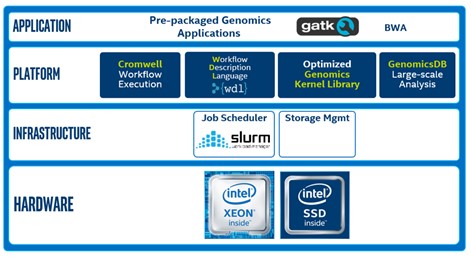
Tuning the Intel® Select Solutions for Genomics Analytics
Software configuration tuning is essential because the operating system and the genomics analysis software were designed for general-purpose applications. The default configurations have not been tuned for the best performance. The following sections provide step-by-step guidance for tuning the Intel® Select Solutions for Genomics Analytics.
Tuning the HPC Cluster
Tuning recommendations for hardware are addressed in this guide: HPC Cluster Tuning on 3rd Generation Intel® Xeon® Scalable Processors at: https://www.intel.com/content/www/us/en/developer/articles/guide/hpc-cluster-tuning-on-3rd-generation-xeon.html
Configuring the Slurm Workload Manager
The Slurm Workload Manager provides a means of scheduling jobs and allocating cluster resources to those jobs. When adding the Slurm workload manager to the HPC Cluster, install the server component on the frontend node.
Installing the Slurm Server on the frontend node
Via Slurm, the PAM (pluggable authentication module) restricts normal user SSH access to compute nodes. This may not be ideal in certain circumstances. Slurm needs a system user for the resource management daemons. The global Slurm configuration file and the cryptographic key that is required by the munge authentication library must be available on every host in the resource management pool. The default configuration supplied with the OpenHPC build of Slurm requires the "slurm" user account. Create a user group and grant unrestricted SSH access. Add the slurm user to this user group as follows:
- Create a user group that is granted unrestricted SSH access:
groupadd sshallowlist
- Add the dedicated Intel® Cluster Checker user to the trust-list (previously called whitelist). This user account should be able to run the Cluster Checker both inside and outside of a resource manager job.
usermod -aG sshallowlist clck
- Create the Slurm user account:
useradd --system --shell=/sbin/nologin slurm
- Install Slurm server packages:
dnf -y install ohpc-slurm-server
- Update the Warewulf files:
wwsh file sync
Summary of the commands
Here are all the commands for this section:
groupadd sshallowlist
usermod -aG sshallowlist clck
useradd --system --shell=/sbin/nologin slurm
dnf -y install ohpc-slurm-server
wwsh file sync
Updating node resource information
Update the Slurm configuration file with the node names of the compute nodes, the properties of their processors, and the Slurm partitions or queues that are associated with your HPC Cluster, including:
- The NodeName tag, or tags, must reflect the names of the compute nodes along with a definition of their respective capabilities.
- The Sockets tag defines the number of sockets in a compute node.
- The CoresPerSocket tag defines the number of CPU cores per socket.
- The ThreadsPerCore tag defines the number of threads that can be executed in parallel on a core.
- The PartitionName tag, or tags, defines the Slurm partitions, or queues, where compute names are assigned. Users use these tags to access these resources. The name of the partition will be visible to the user.
- The Nodes argument defines the compute nodes that belong to a given partition.
- The Default argument defines which partition is the default partition. The default partition is the partition that is used when a user doesn't explicitly specify a partition.
Complete the following tasks to create a Slurm configuration file for a cluster based on the Bill of Materials for this Reference Design:
- Use the openHPC template to create a new Slurm configuration file.
- Add the host name of the frontend host as the ControlMachine to the Slurm configuration file.
- Ensure that the SLURM control daemon will make a node available again when the node registers with a valid configuration after being DOWN
- Update the NodeName definition to reflect the hardware capabilities
- Update the PartitionName definition
Step 1: Create
To create a new SLURM config file, copy the template for the openHPC SLURM config file:
cp /etc/slurm/slurm.conf.ohpc /etc/slurm/slurm.conf
Step 2: Update the configuration file
Open the /etc/slurm/slurm.conf file and make the following changes:
- Locate and update the line beginning with "ControlMachine" to:
ControlMachine=frontend
- The SLURM configuration that ships with OpenHPC has a default set-up. The SLURM control daemon will only make a node available after it goes into the DOWN state if the node was in the DOWN state because it was non-responsive. You can refer to the documentation on the ReturnToService configuration option in: https://slurm.schedmd.com/slurm.conf.html
This configuration is reasonable for a large cluster under constant supervision by a system administrator. For a smaller cluster or a cluster that is not under constant supervision by a system administrator, a different configuration is preferable. SLURM should make a compute node available again when a node with a valid configuration registers with the SLURM control daemon. To enable this type of return to service, locate this line:
ReturnToService=1
Replace it with:
ReturnToService=2
- The SLURM configuration that ships with OpenHPC permits sharing a compute node if the specified resource requirements allow it. Adjust this so that jobs can be scheduled based on CPU and memory requirements.
Locate the line:
SelectType=select/cons_tres
Replace it with the following. Take care to drop the “t” from tres and use the setting: select/cons_res:
SelectType=select/cons_res
Locate the line:
SelectTypeParameters=CR_Core
Replace it with:
SelectTypeParameters=CR_Core_Memory
- Update the node information to reflect the cluster configuration. Locate the NodeName tag. For compute nodes based on 3rd Generation Intel® Xeon® Scalable Gold 6348 processors, replace the existing line with the following line:
NodeName=c[01-04] Sockets=2 CoresPerSocket=28 ThreadsPerCore=2RealMemory=480000 state=UNKNOWN
When configured for use with Cromwell, bwa,all and haplo will be used to provide improved performance. Locate the PartitionName tag and replace the existing line with the following lines.
PartitionName=xeon Nodes=c[01-04] Priority=10000 default=YES MaxTime=24:00:00 State=UP
PartitionName=bwa Nodes=c[01-04] Priority=4000 Default=NO MaxTime=24:00:00 State=UP
PartitionName=all Nodes=c[01-04] Priority=6000 Default=NO MaxTime=24:00:00 State=UP
PartitionName=haplo Nodes=c[01-04] Priority=5000 default=NO MaxTime=24:00:00 State=UP
- The openHPC slurm.conf example has a typo that prevents slurmctld and slurmd from starting. Fix that typo by locating the following line and using a “#” symbol to make it a comment:
Caution: A second line starting with JobCompType exists. DO NOT change that line!
Locate this line:
JobCompType=jobcomp/none
Add the “#” in front to comment it, so that it reads as follows:
#JobCompType=jobcomp/none
- Save and close the file.
Step 3: Import
Import the new configuration files into Warewulf:
wwsh -y file import /etc/slurm/slurm.conf
Step 4: Update the provision configuration file
Update the /etc/warewulf/defaults/provision.conf file:
- Open the /etc/warewulf/defaults/provision.conf file and located the line starting with:
files = dynamic_hosts, passwd, group ...
Note: Do not change any part of the existing line. Append the following to the end of the line. Remember to include a comma at the beginning of the added text.
, slurm.conf, munge.key
- Save and close the file.
Step 5: Enable controller services
Enable MUNGE and Slurm controller services on the frontend node:
syssystemctl enable slurmctld.service
Summary of the commands
Here are all the commands for this section:
cp /etc/slurm/slurm.conf.ohpc /etc/slurm/slurm.conf
sed -i "s/^\(NodeName.*\)/#\1/" /etc/slurm/slurm.conf
echo "NodeName=${compute_prefix}[${first_node}-${last_node}] Sockets=${num_sockets} \
CoresPerSocket=${num_cores} ThreadsPerCore=2 State=UNKNOWN" >> /etc/slurm/slurm.conf
sed -i "s/ControlMachine=.*/ControlMachine=${frontend_name}/" /etc/slurm/slurm.conf
sed -i "s/^\(PartitionName.*\)/#\1/" /etc/slurm/slurm.conf
sed -i "s/^\(ReturnToService.*\)/#\1\nReturnToService=2/" /etc/slurm/slurm.conf
sed -i "s/^\(SelectTypeParameters=.*\)/#\1/" /etc/slurm/slurm.conf
sed -i "s/^\(JobCompType=jobcomp\/none\)/#\1/" /etc/slurm/slurm.conf
cat >> /etc/slurm/slurm.conf << EOFslurm
PartitionName=xeon Nodes=${compute_prefix}[${first_node}-${last_node}] Default=YES MaxTime=24:00:00 State=UP
PartitionName=cluster Nodes=${compute_prefix}[${first_node}-${last_node}] Default=NO MaxTime=24:00:00 \
State=UP
EOFslurm
wwsh -y file import /etc/slurm/slurm.conf
sed -i "s/^files\(.*\)/files\1, slurm.conf, munge.key/" /etc/warewulf/defaults/provision.conf
syssystemctl enable slurmctld.service
Installing the Slurm Client into the compute node image
- Add Slurm client support to the compute node image:
dnf -y --installroot=$CHROOT install ohpc-slurm-client
- Create a munge.key file in the compute node image. It will be replaced with the latest copy at node boot time.
\cp -fa /etc/munge/munge.key $CHROOT/etc/munge/munge.key
- Update the initial Slurm configuration on the compute nodes. The Warewulf synchronization mechanism does not synchronize during early boot.
\cp -fa /etc/slurm/slurm.conf $CHROOT/etc/slurm/
- Enable the Munge and Slurm client services:
systemctl --root=$CHROOT enable munge.service
- Allow unrestricted SSH access for the root user and the users in the sshallowlist group. Other users will be subject to SSH restrictions. Open $CHROOT/etc/security/access.conf and append the following lines, in order, to the end of the file.
+ : root : ALL
+ : sshallowlist : ALL
- : ALL : ALL
- Enable SSH control via the Slurm workload manager. Enabling PAM in the chroot environment limits SSH access to only those nodes where the user has active jobs. First, open $CHROOT/etc/pam.d/sshd and append the following lines, in order, to the end of the file:
account sufficient pam_access.so
account required pam_slurm.so
Then, save and close the file.
echo "account sufficient pam_access.so" >> $CHROOT/etc/pam.d/sshd
- Update the VNFS image:
wwvnfs --chroot $CHROOT –hybridize
Summary of the commands
Here are all the commands for this section:
echo "+ : root : ALL">>$CHROOT/etc/security/access.conf
echo "+ : sshallowlist : ALL">>$CHROOT/etc/security/access.conf echo "- : ALL : ALL">>$CHROOT/etc/security/access.conf
echo "account sufficient pam_access.so" >> $CHROOT/etc/pam.d/sshd
wwvnfs --chroot $CHROOT –hybridize
Optional – Installing the Slurm client on the frontend node
If you also want to use the cluster frontend node for compute tasks that are scheduled via Slurm, enable it as follows:
- Add Slurm client package to the frontend node:
dnf -y install ohpc-slurm-client
- Add the frontend node as a compute node to the SLURM. Open /etc/slurm/slurm.conf and locate the NodeName tag. For compute nodes based on 3rd Generation Intel® Xeon® Scalable Gold 6348 processors, replace the existing line with the following line. This should be one single line:
NodeName=frontend,c[01-04] Sockets=2 CoresPerSocket=28 ThreadsPerCore=2 State=UNKNOWN
Locate the PartitionName tag and replace the existing line with these two lines as two single lines:
PartitionName=xeon Nodes=frontend,c[01-04] Default=YES MaxTime=24:00:00 State=UP PartitionName=cluster Nodes=frontend,c[01-04] Default=NO MaxTime=24:00:00 State=UP
- Restart the Slurm control daemon:
systemctl restart slurmctld
- Enable and start the Slurm daemon on the frontend node:
systemctl enable --now slurmd
- Update the initial Slurm configuration on the compute nodes:
\cp -fa /etc/slurm/slurm.conf $CHROOT/etc/slurm/
- Update the VNFS image:
wwvnfs --chroot $CHROOT --hybridize
Summary of the commands
Here are all the commands in this section:
sed -i "s/NodeName=/NodeName=frontend,/" /etc/slurm/slurm.conf
systemctl restart slurmctld
systemctl enable --now slurmd
\cp -fa /etc/slurm/slurm.conf $CHROOT/etc/slurm/
wwvnfs --chroot $CHROOT --hybridize
Finalizing the Slurm configuration
- The resource allocation in Slurm version 20.11.X has been changed. This has direct implications for certain types of MPI based jobs. For details, see the Slurm release notes at: https://slurm.schedmd.com/archive/slurm-20.11.6/news.html
In order to allow applications to use the resource allocation method from Slurm version 20.02.X and earlier, set the "SLURM_OVERLAP" environment variable on the frontend node to allow all users:
cat >> /etc/environment << EOFSLURM export SLURM_OVERLAP=1 EOFSLURM
- Re-start the Munge and Slurm controller services on the frontend node:
systemctl restart munge
- Reboot the compute nodes:
pdsh -w c[01-XX] reboot
- Ensure that the compute nodes are available in Slurm after they have been re- provisioned. In order to ensure that the compute nodes are available for resource allocation in Slurm, they need to be put into the "idle" state.
scontrol reconfig
scontrol update NodeName=c[01-XX] State=Idle
- Check the Slurm status. All nodes should have the state "idle".
[root@frontend ~]# sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
xeon* up 1-00:00:00 4 idle c[01-04]
cluster up 1-00:00:00 4 idle c[01-04]
Summary of the commands
Here are all the commands in this section:
cat >> /etc/environment << EOFSLURM export SLURM_OVERLAP=1 EOFSLURM
systemctl restart munge
pdsh -w c[01-XX] reboot
scontrol reconfig
scontrol update NodeName=c[01-XX] State=Idle
[root@frontend ~]# sinfo
Running a test
Once the resource manager is in production, normal users should be able to run jobs. Test this by creating a "test" user with standard privileges. For example, this user will not be allowed to use SSH to interact with the compute nodes outside of a Slurm job. Compile and execute a "hello world" application interactively through the resource manager. Note the use of srun for parallel job execution because it summarizes the underlying, native job launch mechanism.
- Add a "test" user:
useradd -m test
- Create a password for the user:
passwd test
- Synchronize the files with the Warewulf database. Note: The synchronization to the compute nodes can take several minutes.
wwsh file resync
- Switch to the "test" user account:
su – test
- Source the environment for the MPI implementation that will be used. In this example, the Intel MPI provided by oneAPI was used.
module load oneapi
- Compile the MPI "hello world" example:
mpicc -o test /opt/intel/oneapi/mpi/latest/test/test.c
- Submit the job. Start a Slurm job on all nodes. This should produce output similar to the following:
[test@frontend ~]# srun --mpi=pmi2 --partition=xeon -N 4 -n 4 /home/test/test
Hello world: rank 0 of 4 running on c01.cluster
- When the MPI job rans successfully, exit as the "test" user and continue with the cluster setup:
exit
Installing the genomics software stack
Install the genomics software stack. This software stack is based on the Genome Analysis ToolKit (GATK) suite of tools along with the Cromwell workflow management system.
Checking the setup of the genomics cluster environment
First, check that the cluster setup used to run the Genomics solution is properly configured.
- Make sure that Cromwell users on every node are limited to opening no more than 500,000 files:
pdsh -w frontend,c0[1-4] "su - cromwell -c 'ulimit -n'"
Note: This should be at least 500,000 on every node because workflow settings require many files to be open during runtime.
- Check that the /genomics_local is a mounted filesystem with the correct ownership and permissions. The pdsh command should produce output identical to the one shown, except the order of compute nodes may vary.
[root@frontend ~]# pdsh -w c0[1-4] "stat -c '%A %U:%G %m' /genomics_local"
c01: drwxr-xr-x cromwell:cromwell /genomics_local
c03: drwxr-xr-x cromwell:cromwell /genomics_local
c04: drwxr-xr-x cromwell:cromwell /genomics_local
c02: drwxr-xr-x cromwell:cromwell /genomics_local
Configuring MariaDB
Cromwell uses MariaDB for storing job information. Since Warewulf also uses MariaDB, the database may have been setup according to the HPC Cluster guide during the hardware configuration. If not, follow these instructions to set it up. You will need the MariaDB database administrator password.
- Enable the database service to start automatically at system boot
systemctl enable mariadb.service
- Enable the web server service to start automatically at system boot
systemctl enable httpd.service
- Restart the database service
systemctl restart mariadb
- Restart web services
systemctl restart httpd
- Update the Warewulf database password. This step must be done manually. First, edit this file: /etc/warewulf/database-root.conf Replace the "changeme" password with a password that you choose based on your password policy:
database password = <new_password>
Note: We recommend that this password be different than the password for the root superuser.
6. Save and exit the file.
7. Secure database. MariaDB ships with a tool that allows the administrator to manually setup database access. This includes setting a password for the root user of the database. We recommend that this password be different from the password of the root user of the system and password for the Warewulf database administrator. Please follow your password policy when choosing passwords. Execute the following command and respond to the prompts. We strongly recommend using the default answers for all questions.
/usr/bin/mysql_secure_installation
- Initialize the Warewulf data store system. Enter the database root password when prompted.
wwinit DATASTORE
- Log into the MariaDB server as the database administrator and enter the root password when prompted:
mysql -h localhost -u root -p
Note: If you are unable to log into MariaDB see the Troubleshooting of the genomics software stack section of this guide for suggestions
- Create a new database named “cromwell”:
MariaDB [(none)]> CREATE DATABASE cromwell;
- Create a new MySQL database user. In this example, “cromwell’ is also the password. Change the password to meet your password security policy.
MariaDB [(none)]> CREATE USER 'cromwell'@'localhost' IDENTIFIED BY 'cromwell';
- Grant database permissions to the new user. Replace “cromwell” with your password.
MariaDB [(none)]> GRANT ALL PRIVILEGES ON `cromwell`.* TO 'cromwell'@'localhost';
- Reload and exit:
MariaDB [(none)]> FLUSH PRIVILEGES;
MariaDB [(none)]> exit
Installing the Cromwell Workflow Management System
Install the Cromwell Workflow Management system and configure it to use the local scratch device on the compute nodes.
- In order to install Cromwell, the "sbt" build tool is required. First, install sbt. Add the sbt online repository
dnf config-manager --add-repo https://www.scala-sbt.org/sbt-rpm.repo
Then, install sbt build tool:
dnf -y install sbt
- Download Cromwell from the git repository and create a directory for installing Cromwell:
mkdir -p ${GENOMICS_PATH}/cromwell
Set the owner of that directory to the cromwell user and the cromwell group. Change the ownership permissions in order to allow access.
chmod 775 -R ${GENOMICS_PATH}/Cromwell
chown -R cromwell:cromwell ${GENOMICS_PATH}/Cromwell
Login to the cromwell user account:
su - cromwell
Use git to clone the Cromwell git repository:
cd ${GENOMICS_PATH}/cromwell
git clone https://github.com/broadinstitute/cromwell.git
Since this guide was tested and validated with version 52 of Cromwell, checkout version 52 from the repository:
cd cromwell
git checkout 52
- Configure Cromwell to use the local NVMe disk as scratch space. Open this file for editing:
backend/src/main/scala/cromwell/backend/RuntimeEnvironment.scala
Comment out line 3, so it reads:
//import java.util.UUID
Update lines 23 through 27 as shown below:
val tempPath: String = {
val uuid = UUID.randomUUID().toString
val hash = uuid.substring(0, uuid.indexOf('-')) callRoot.resolve(s"tmp.$hash").pathAsString
}
Add the following text in line 23:
def tempPath: String = "/genomics_local"
Save the file and exit. Then, open this file:
backend/src/main/scala/cromwell/backend/standard/ StandardAsyncExecutionActor.scala
Go to line number 380 to find the following content:
|export _JAVA_OPTIONS=-Djava.io.tmpdir="$$tmpDir"
|export TMPDIR="$$tmpDir"
Replace those two lines with the following text:
|mkdir -p $$tmpDir/tmp.$$$$
|export _JAVA_OPTIONS=-Djava.io.tmpdir="$$tmpDir/tmp.$$$$"
|export TMPDIR="$$tmpDir/tmp.$$$$
Save the file and exit.
- Build Cromwell with the patches:
sbt clean
- When the build is successful, move the new jar file into the ${GENOMICS_PATH}/cromwell directory
cp server/target/scala-2.12/cromwell-52-*-SNAP.jar \
${GENOMICS_PATH}/cromwell/cromwell-52-fix.jar
Summary of the commands
Here are all the commands for this section
sed -i "s/^\(import\ java.util.UUID\)/\/\/\1/" \
backend/src/main/scala/cromwell/backend/RuntimeEnvironment.scala
sed -i '23,27d' \ backend/src/main/scala/cromwell/backend/RuntimeEnvironment.scala
sed -i '23i \ \ \ \ \ \ def tempPath: String = \ "/genomics_local \ "' \ backend/src/main/scala/cromwell/backend/RuntimeEnvironment.scala
sed 's/\(\s*|export _JAVA_OPTIONS.*\) \ "/\ \ \ \ \ \ \ \ |mkdir -p \$\$tmpDir\/tmp\.\$\$\$\$\n\1 \ "/' \ backend/src/main/scala/cromwell/backend/standard/StandardAsyncExecutionActor.scala
sed 's/\(\s*|export _JAVA_OPTIONS.*tmpDir\) \ "/\1\/tmp\.\$\$\$\$ \ "/' \ backend/src/main/scala/cromwell/backend/standard/StandardAsyncExecutionActor.scala
sed 's/\(\s*|export TMPDIR=.*tmpDir\) \ "/\1\/tmp\.\$\$\$\$ \ "/' \ backend/src/main/scala/cromwell/backend/standard/StandardAsyncExecutionActor.scala
Edit backend/src/main/scala/cromwell/backend/standard/ StandardAsyncExecutionActor.scala
Replace line 380-381 with:
|mkdir -p $$tmpDir/tmp.$$$$
|export _JAVA_OPTIONS=-Djava.io.tmpdir="$$tmpDir/tmp.$$$$"
|export TMPDIR="$$tmpDir/tmp.$$$$
sbt clean
cp server/target/scala-2.12/cromwell-52-*-SNAP.jar ${GENOMICS_PATH}/cromwell/cromwell-52-fix.jar
Configuring the execution environment for Cromwell
Configure the Cromwell execution environment. Use the default configuration file as a starting point and change it to reflect your needs.
- Configure MariaDB as the database for Cromwell to use
- Add the SLURM backends, so Cromwell can schedule jobs using SLURM
Step-by-step instructions
- First, download the default Cromwell reference.conf configuration file.
wget https://raw.githubusercontent.com/broadinstitute/cromwell/52_hotfix/core/\
- Add MariaDB as the database for Cromwell to use. Open this file: ${GENOMICS_PATH}/cromwell/reference.conf and add the following lines at the end of the file:
database {
profile = "slick.jdbc.MySQLProfile$" db {
driver = "com.mysql.cj.jdbc.Driver"
url = "jdbc:mysql://localhost/cromwell?rewriteBatchedStatements=true&serverTimezone=UTC" user = "cromwell"
password = "cromwell" connectionTimeout = 5000
}
}
- Add SLURM as the backend for Cromwell. Open this file: ${GENOMICS_PATH}/cromwell/reference.conf then go to line 479 to find:
default = "Local"
Change "Local" to "SLURM"
default = "SLURM"
Remove the content on lines 480 through 484
providers {
Local {
# Modifying temp directory to write to local disk temporary-directory = "$(/genomics_local/)"
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory"
config {
include required(classpath("reference_local_provider_config.inc.conf"))
Now add the following text after line 479, the line that has default = "SLURM".
Note: Ensure that the lines that show line-breaks in this document are, in fact, single lines in reference.conf
providers { SLURM {
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory" config {
root = "cromwell-slurm-exec" runtime-attributes = """
Int runtime_minutes = 600 Int cpu = 2
Int memory_mb = 1024 String queue = "all"
String? docker """
submit = """
sbatch -J ${job_name} -D ${cwd} -o ${out} -e ${err} -t ${runtime_minutes} -p ${queue} ${"-c " + cpu} --mem ${memory_mb} --wrap "/bin/bash ${script}"
"""
kill = "scancel ${job_id}"
check-alive = "squeue -j ${job_id}"
job-id-regex = "Submitted batch job (\\d+).*"
}
}
SLURM-BWA {
temporary-directory = "$(/genomics_local/)"
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory" config {
root = "cromwell-slurm-exec" runtime-attributes = """
Int runtime_minutes = 600 Int cpu = 2
Int memory_mb = 1024 String queue = "bwa"
String? docker """
submit = """
sbatch -J ${job_name} -D ${cwd} -o ${out} -e ${err} -t ${runtime_minutes} -p ${queue} ${"-c " + cpu} --mem ${memory_mb} --wrap "/bin/bash ${script}"
"""
kill = "scancel ${job_id}"
check-alive = "squeue -j ${job_id}"
job-id-regex = "Submitted batch job (\\d+).*"
}
}
SLURM-HAPLO {
temporary-directory = "$(/genomics_local/)"
actor-factory = "cromwell.backend.impl.sfs.config.ConfigBackendLifecycleActorFactory" config {
root = "cromwell-slurm-exec" runtime-attributes = """
Int runtime_minutes = 600 Int cpu = 2
Int memory_mb = 1024 String queue = "haplo"
String? docker """
submit = """
sbatch -J ${job_name} -D ${cwd} -o ${out} -e ${err} -t ${runtime_minutes} -p ${queue} ${"-c " + cpu} --mem ${memory_mb} --wrap "/bin/bash ${script}"
"""
kill = "scancel ${job_id}"
check-alive = "squeue -j ${job_id}"
job-id-regex = "Submitted batch job (\\d+).*"
Summary of the Commands
Here are all the commands in this section:
sed -i '$ a database {' reference.conf
sed -i '$ a \ \ profile = \"slick.jdbc.MySQLProfile$\"' reference.conf sed -i '$ a \ \ db {' reference.conf
sed -i '$ a \ \ \ \ driver = \"com.mysql.cj.jdbc.Driver\"' reference.conf
sed -i '$ a \ \ \ \ url = \"jdbc:mysql:\/\/localhost\/cromwell\?rewriteBatched\ Statements=true&serverTimezone=UTC\"' reference.conf
sed -i '$ a \ \ \ \ user = \"cromwell\"' reference.conf
sed -i '$ a \ \ \ \ password = \"cromwell\"' reference.conf sed -i '$ a \ \ \ \ connectionTimeout = 5000' reference.conf sed -i '$ a \ \ }' reference.conf
sed -i '$ a }' reference.conf
sed -i '479,484d' reference.conf
sed -i "479i \ \ \ \ \ \ \ \ job-id-regex\ =\ \"Submitted\ batch\ job\ (\\\\\\\\d+).*\"" \ reference.conf
sed -i "479i \ \ \ \ \ \ \ \ check-alive\ =\ \"squeue\ -j\ \${job_id}\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ kill\ =\ \"scancel\ \${job_id}\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ \"\"\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ sbatch\ -J\ \${job_name}\ -D\ \${cwd}\ -o\ \${out}\ -e\ \${err}\ \
-t\ \${runtime_minutes}\ -p\ \${queue}\ \${\"-c\ \"\ +\ cpu}\ --mem\ \${memory_mb}\ \
--wrap\ \"\/bin\/bash\ \${script}\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ submit\ =\ \"\"\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ \"\"\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ String?\ docker" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ String\ queue\ =\ \"haplo\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ Int\ memory_mb\ =\ 1024" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ Int\ cpu\ =\ 2" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ Int\ runtime_minutes\ =\ 600" reference.conf sed -i "479i \ \ \ \ \ \ \ \ runtime-attributes\ =\ \"\"\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ root\ =\ \"cromwell-slurm-exec\"" reference.conf sed -i "479i \ \ \ \ \ \ config\ {" reference.conf
sed -i "479i \ \ \ \ \ \ actor-factory\ =\ \"cromwell.backend.impl.sfs.config.\ ConfigBackendLifecycleActorFactory\"" reference.conf
sed -i "479i \ \ \ \ \ \ temporary-directory\ =\ \"\$(\/genomics_local\/)\"" reference.conf sed -i "479i \ \ \ \ SLURM-HAPLO\ {" reference.conf
sed -i "479i \ \ \ \ }\ \ " reference.conf sed -i "479i \ \ \ \ \ \ }" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ job-id-regex\ =\ \"Submitted\ batch\ job\ (\\\\\\\\d+).*\"" \ reference.conf
sed -i "479i \ \ \ \ \ \ \ \ check-alive\ =\ \"squeue\ -j\ \${job_id}\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ kill\ =\ \"scancel\ \${job_id}\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ \"\"\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ sbatch\ -J\ \${job_name}\ -D\ \${cwd}\ -o\ \${out}\ -e\ \${err}\ \
-t\ \${runtime_minutes}\ -p\ \${queue}\ \${\"-c\ \"\ +\ cpu}\ --mem\ \${memory_mb}\ \
--wrap\ \"\/bin\/bash\ \${script}\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ submit\ =\ \"\"\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ \"\"\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ String?\ docker" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ String\ queue\ =\ \"bwa\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ Int\ memory_mb\ =\ 1024" reference.conf sed -i "479i \ \ \ \ \ \ \ \ Int\ cpu\ =\ 2" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ Int\ runtime_minutes\ =\ 600" reference.conf sed -i "479i \ \ \ \ \ \ \ \ runtime-attributes\ =\ \"\"\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ root\ =\ \"cromwell-slurm-exec\"" reference.conf sed -i "479i \ \ \ \ \ \ config\ {" reference.conf
sed -i "479i \ \ \ \ \ \ actor-factory\ =\ \"cromwell.backend.impl.sfs.config.\ ConfigBackendLifecycleActorFactory\"" reference.conf
sed -i "479i \ \ \ \ \ \ temporary-directory\ =\ \"\$(\/genomics_local\/)\"" reference.conf sed -i "479i \ \ \ \ SLURM-BWA\ {" reference.conf
sed -i "479i \ \ \ \ }" reference.conf
sed -i "479i \ \ \ \ \ \ }\ \ " reference.conf
sed -i "479i \ \ \ \ \ \ \ \ job-id-regex\ =\ \"Submitted\ batch\ job\ (\\\\\\\\d+).*\"" \ reference.conf
sed -i "479i \ \ \ \ \ \ \ \ check-alive\ =\ \"squeue\ -j\ \${job_id}\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ kill\ =\ \"scancel\ \${job_id}\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ \"\"\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ sbatch\ -J\ \${job_name}\ -D\ \${cwd}\ -o\ \${out}\ -e\ \${err}\ \
-t\ \${runtime_minutes}\ -p\ \${queue}\ \${\"-c\ \"\ +\ cpu}\ --mem\ \${memory_mb}\ \
--wrap\ \"\/bin\/bash\ \${script}\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ submit\ =\ \"\"\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ \"\"\"" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ String?\ docker" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ String\ queue\ =\ \"all\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ Int\ memory_mb\ =\ 1024" reference.conf sed -i "479i \ \ \ \ \ \ \ \ Int\ cpu\ =\ 2" reference.conf
sed -i "479i \ \ \ \ \ \ \ \ Int\ runtime_minutes\ =\ 600" reference.conf sed -i "479i \ \ \ \ \ \ \ \ runtime-attributes\ =\ \"\"\"" reference.conf sed -i "479i \ \ \ \ \ \ \ \ root\ =\ \"cromwell-slurm-exec\"" reference.conf sed -i "479i \ \ \ \ \ \ config\ {" reference.conf
sed -i "479i \ \ \ \ \ \ actor-factory\ =\ \"cromwell.backend.impl.sfs.config.\ ConfigBackendLifecycleActorFactory\"" reference.conf
sed -i "479i \ \ \ \ \ \ temporary-directory\ =\ \"\$(\/genomics_local\/)\"" reference.conf
sed -i "479i \ \ \ \ \ \ #\ Modifying\ temp\ directory\ to\ write\ to\ local\ disk" reference.conf sed -i "479i \ \ \ \ SLURM\ {" reference.conf
sed -i "479i \ \ providers\ {" reference.conf
sed -i "479i \ \ default\ =\ \"SLURM\"" reference.conf
Validating the Cromwell configuration
Validate that the Cromwell installation is working properly by executing a simple smoke test.
- Change to the "cromwell" user
su - cromwell
- Change to the Cromwell directory
cd ${GENOMICS_PATH}/cromwell
- Start the Cromwell server as a detached process in the background. By default the Cromwell server will be accessible via port 8000. The cromwell.log file will contain the messages the Cromwell server generates during execution.
nohup java -jar -Dconfig.file=reference.conf cromwell-52-fix.jar server 2>&1 >>cromwell.log &
- Confirm that the Cromwell server is running by checking the process list by using the "ps" command:
- Note: The output of the "ps" command will output the PID and other information that we are not concerned with. We are using <…> as a placeholder for that information.*
[cromwell@frontend cromwell]$ ps aux | grep cromwell | grep server
cromwell <PID> <…> java -jar -Dconfig.file=reference.conf cromwell-52-fix.jar server
Note: In order to stop the Cromwell server, use the "kill -9 " command where is the process ID running Cromwell.
- Now that the Cromwell server process is running, execute an example workflow. This workflow was designed using the Workflow Description Language (WDL). It will ensure that Cromwell is configured correctly with the job scheduler. Navigate to your working directory:
cd ${GENOMICS_PATH}/cromwell/
Open a new file named HelloWorld.wdl and add the following text:
task hello {
String name command {
echo 'Hello ${name}!'
}
output {
File response = stdout()
}
runtime {
memory: "2MB" disk: "2MB"
Save the file and exit. Open a new file called: HelloWorld.json and add the following text to that file:
{"helloWorld.hello.name": "World"}
Save the file and exit. We can now submit the workflow to the Cromwell server using the WDL and JSON input files that were just created:
curl -v http://127.0.0.1:8000/api/workflows/v1 -F \ workflowSource=@${GENOMICS_PATH}/cromwell/HelloWorld.wdl -F \ workflowInputs=@${GENOMICS_PATH}/cromwell/HelloWorld.json
Use the job id provided by the output to check the status of the workflow:
curl -v http://127.0.0.1:8000/api/workflows/v1/<id>/status
#After a few minutes, the above command should return a “succeeded” message.
Summary of the commands
Here are all the commands in this section:
su – Cromwell
cd ${GENOMICS_PATH}/cromwell
nohup java -jar -Dconfig.file=reference.conf cromwell-52-fix.jar server 2>&1 >>cromwell.log &
[cromwell@frontend cromwell]$ ps aux | grep cromwell | grep server
cromwell <PID> <…> java -jar -Dconfig.file=reference.conf cromwell-52-fix.jar server
cd ${GENOMICS_PATH}/cromwell/
cat >> HelloWorld.wdl << EOFwdl task hello {
String name command {
echo 'Hello ${name}!'
}
output {
File response = stdout()
}
runtime {
memory: "2MB" disk: "2MB"
}
}
workflow helloWorld { call hello
}
EOFwdl
echo {\"helloWorld.hello.name\":\ "World\"}" >> HelloWorld.json
curl -v http://127.0.0.1:8000/api/workflows/v1 -F \ workflowSource=@${GENOMICS_PATH}/cromwell/HelloWorld.wdl -F \ workflowInputs=@${GENOMICS_PATH}/cromwell/HelloWorld.json
curl -v http://127.0.0.1:8000/api/workflows/v1/<id>/status
#After a few minutes, the above command should return a “succeeded” message.
Installing the Burrows-Wheeler Aligner (BWA)
BWA is a software package for mapping low-divergent sequences against a large reference genome, such as the human genome. In order to install the Burrows- Wheeler Aligner requires you must compile it.
- Login to the cromwell user account:
su - cromwell
- Create the tools subdirectory in the GENOMICS_PATH directory and enter that directory:
mkdir ${GENOMICS_PATH}/tools
- Download the recommended version of the BWA software package:
wget https://github.com/lh3/bwa/releases/download/v0.7.17/bwa-0.7.17.tar.bz2
- Unpack the tarball:
tar -xjf bwa-0.7.17.tar.bz2
- Compile BWA:
cd bwa-0.7.17
- Make a symlink:
cd ${GENOMICS_PATH}/tools
Summary of the commands
Here are all the commands in this section:
su - cromwell
mkdir ${GENOMICS_PATH}/tools
wget https://github.com/lh3/bwa/releases/download/v0.7.17/bwa-0.7.17.tar.bz2
tar -xjf bwa-0.7.17.tar.bz2
cd bwa-0.7.17
cd ${GENOMICS_PATH}/tools
Installing the Genome Analysis ToolKit (GATK)
The Genome Analysis ToolKit (GATK) is the industry standard for identifying SNPs and indels in germline DNA and RNAseq data. It is a collection of command-line tools for analyzing high-throughput sequencing data with a primary focus on variant discovery. The tools can be used individually or chained together into complete workflows. The Broad Institute provides end-to-end workflows, called GATK Best Practices, tailored for specific use cases. The Intel® Genomics Kernel Library with AVX-512 performance improvements is directly integrated into GATK.
- Navigate to the staging folder:
cd ${GENOMICS_PATH}/tools
- Download the latest version of GATK:
wget \
https://github.com/broadinstitute/gatk/releases/download/4.1.9.0/gatk-4.1.9.0.zip
- Unpack the zip file:
unzip gatk-4.1.9.0.zip
- Add the PATH to the default user environment:
echo "export PATH=\${GENOMICS_PATH}/tools/gatk-4.1.9.0:\$PATH" >> /etc/bashrc
- Make tools symlink:
cd ${GENOMICS_PATH}/tools
ln -s gatk-4.1.9.0 gatk
ln -s gatk-4.1.9.0/gatk-package-4.1.9.0-local.jar gatk-jar
Summary of the commands
Here are all the commands in this section:
cd ${GENOMICS_PATH}/tools
wget \
https://github.com/broadinstitute/gatk/releases/download/4.1.9.0/gatk-4.1.9.0.zip
unzip gatk-4.1.9.0.zip
echo "export PATH=\${GENOMICS_PATH}/tools/gatk-4.1.9.0:\$PATH" >> /etc/bashrc
cd ${GENOMICS_PATH}/tools
ln -s gatk-4.1.9.0 gatk
ln -s gatk-4.1.9.0/gatk-package-4.1.9.0-local.jar gatk-jar
Installing Picard
- When logged on as user, Cromwell, navigate to the staging folder:
cd ${GENOMICS_PATH}/tools
- Download the recommended version of Picard
wget https://github.com/broadinstitute/picard/releases/download/2.23.8/picard.jar
Summary of the commands
Here are all the commands in this section:
cd ${GENOMICS_PATH}/tools
wget https://github.com/broadinstitute/picard/releases/download/2.23.8/picard.jar
Installing Samtools
- Change to the tools directory
cd ${GENOMICS_PATH}/tools
- Download the recommended version of the SAMtools software:
wget https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
- Unpack the tarball:
tar -xjf samtools-1.9.tar.bz2
- Compile and install Samtools:
cd samtools-1.9
./configure -prefix=${GENOMICS_PATH}/tools
make
- Make a symlink:
${GENOMICS_PATH}/tools
ln -s samtools-1.9 samtools
##### Summary of the commands
Here are all the commands in this section:
cd ${GENOMICS_PATH}/tools
wget https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
tar -xjf samtools-1.9.tar.bz2
cd samtools-1.9
./configure -prefix=${GENOMICS_PATH}/tools
make
${GENOMICS_PATH}/tools
ln -s samtools-1.9 samtools
Installing VerifyBamID2
You must build the project from the source code. See the details at: https:// github.com/Griffan/VerifyBamID
- Navigate to the tools folder:
cd ${GENOMICS_PATH}/tools
- Create the file build_verify.sh and add the following contents:
#https://github.com/broadinstitute/warp/tree/cec97750e3819fd88ba382534aaede8e05ec52df/dockers/broad/ VerifyBamId
cd $GENOMICS_PATH/tools
VERIFY_BAM_ID_COMMIT="c1cba76e979904eb69c31520a0d7f5be63c72253" GIT_HASH=$VERIFY_BAM_ID_COMMIT
HTS_INCLUDE_DIRS=$GENOMICS_PATH/tools/samtools-1.9/htslib-1.9/ HTS_LIBRARIES=$GENOMICS_PATH/tools/samtools-1.9/htslib-1.9/libhts.a
wget -nc https://github.com/Griffan/VerifyBamID/archive/$GIT_HASH.zip && \ unzip -o $GIT_HASH.zip && \
cd VerifyBamID-$GIT_HASH && \ mkdir build && \
cd build && \
echo cmake -DHTS_INCLUDE_DIRS=$HTS_INCLUDE_DIRS -DHTS_LIBRARIES=$HTS_LIBRARIES .. && \
CC=$(which gcc) CXX=$(which g++) \
cmake -DHTS_INCLUDE_DIRS=$HTS_INCLUDE_DIRS -DHTS_LIBRARIES=$HTS_LIBRARIES .. && \
make && \
make test && \ cd ../../
mv $GENOMICS_PATH/tools/VerifyBamID-$GIT_HASH $GENOMICS_PATH/tools/VerifyBamID && \ rm -rf $GENOMICS_PATH/tools/$GIT_HASH.zip $GENOMICS_PATH/tools/VerifyBamID-$GIT_HASH
- Run the new script to build and install VerifyBamID2:
sh ./build_verify.sh
Using the 20K Throughput Run to verify your configuration
The 20K Sample Test provides a quick end-to-end smoke test. It runs a Broad Best Practices Workflow with a short input dataset. A single Whole Genome Sequence (WGS) can take hours to run. Using the recommended hardware, this test should complete in 30-40 minutes.
- Login to the cromwell user account:
su - cromwell
- Navigate to the cromwell installation folder:
cd ${GENOMICS_PATH}/cromwell
- Clone the workflow repo (as a user). Configure Git proxy settings as needed for your environment.
git clone https://github.com/Intel-HLS/BIGstack.git
- Review the values in the "configure" values for proper DATA and TOOL paths on your system. DATA should be a filesystem all worker nodes can reach and have at least 2TB of free space. The benchmark Step2 will download data from the Broad Institute and configure the proxy settings as setup in configure.
vim configure
- Configure the test based on the detail in the "configure" file:
./step01_Configure_20k_Throughput-run.sh
- Download data in the DATA_PATH provided in the configure file:
./step02_Download_20k_Data_Throughput-run.sh
- Run the Throughput benchmark:
./step03_Cromwell_Run_20k_Throughput-run.sh
- Monitor the workflow for the Workflow status - Failed, Succeeded, Running:
./step04_Cromwell_Monitor_Single_Sample_20k_Workflow.sh
- To view the final output of 20k Throughput run, execute
step05_Single_Sample_20k_Workflow_Output.sh
./step05_Single_Sample_20k_Workflow_Output.sh
Total Elapsed Time for 64 workflows: 'X' minutes: 'Y' seconds
Average Elapsed Time for Mark Duplicates: 'X.YZ' minutes
Troubleshooting the genomics software stack
The following section describes steps for troubleshooting issues that may arise during the installation of the genomics software stack.
Troubleshooting failure to log into MariaDB
- Stop MariaDB:
systemctl stop mariadb
- Start a safe session and login:
mysqld_safe --skip-grant-tables --skip-networking & mysql -u root
- Set a new password for root user or change password as desired:
MariaDB [(none)]> FLUSH PRIVILEGES;
MariaDB [(none)]> SET PASSWORD FOR 'root'@'localhost' = PASSWORD('password'); MariaDB [(none)]> EXIT;
- Cleanup and restart MariaDB:
kill `cat /var/run/mariadb/mariadb.pid` systemctl start mariadb
- Log into the MariaDB server as the database administrator. Enter the root password when prompted and resume the setup process:
mysql -h localhost -u root -p
Installing optional components
The Intel® System Configuration Utility, Save and Restore System Configuration Utility (syscfg), is a command-line utility that can be used to display or set several BIOS and management firmware settings.
Conclusion
This guide contains recommendations for tuning software used in the Intel® Select Solutions for Genomics Analytics on the 3rd Generation Intel® Xeon® Scalable Processors Based Platform. HPC Cluster Tuning on 3rd Generation Intel® Xeon® Scalable Processors describes the hardware configuration.
Feedback
We value your feedback. If you have comments (positive or negative) on this guide or are seeking something that is not part of this guide, please reach out and let us know what you think.